High-Throughput Binding Affinity Calculations (HTBAC)
NSF Award 1713749
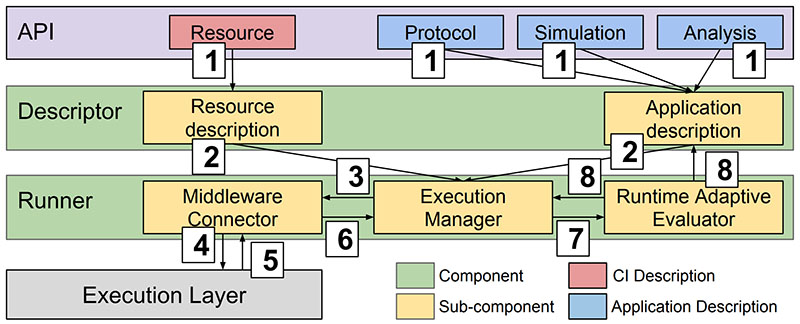
This is a collaborative project that studies a wide range of cancer drugs and candidate ligands. The goal is to support personalized clinical decision-making based on genome sequencing and drug discovery. We use ensemble-based, high-performance computing methods, running automated workflows at unprecedented scales. We developed the High-Throughput Binding Affinity Calculator (HTBAC), integrating of the Binding Affinity Calculator (BAC), developed by the Centre for Computational Science at UCL, with RADICAL-Cybertools, developed by the RADICAL Laboratory at Rutgers University. HTBAC enables rapid, accurate, scalable, reliable and adaptive free energy-based binding affinity calculations, separating the definition of a free energy protocols from its distributed execution. Adaptivity allows users to change the course of execution during runtime, favoring interesting simulations and drug candidates.